![]()
Practice Essentials
Mitral valve prolapse (MVP) is the most common valvular abnormality, affecting approximately 2-6% of the population in the United States. MVP usually has a benign course, but it occasionally leads to serious complications, including clinically significant mitral regurgitation, infective endocarditis, sudden cardiac death, and cerebrovascular ischemic events.
Essential update: More accurate imaging of mitral valve prolapse with 3D echocardiography
Real-time 3-dimensional transesophageal echocardiography (3D TEE) is superior to 2-dimensional TEE for quantifying MVP in patients with severe mitral regurgitation, according to a study of 102 patients with severe mitral regurgitation due to MVP, mitral valve flail, or both. Compared with 3D TEE, 2D TEE underestimated the width of the prolapse and the leaflet gap. In addition, because of the complicated anatomy of the mitral valve, 2D imaging could not detect the largest prolapse gap and width.[1]
Signs and Symptoms
Most patients with MVP are asymptomatic. Symptoms are related to one of the following:
Autonomic dysfunctionProgression of mitral regurgitationAn associated complication (ie, stroke, endocarditis, or arrhythmia)
Symptoms related to autonomic dysfunction are usually associated with genetically inherited MVP and include the following:
AnxietyPanic attacksArrhythmiasExercise intolerancePalpitationsAtypical chest painFatigueOrthostasisSyncope or presyncopeNeuropsychiatric symptoms
Symptoms related to progression of mitral regurgitation include the following:
FatigueDyspneaExercise intoleranceOrthopneaParoxysmal nocturnal dyspnea (PND)Progressive signs of chronic heart failure (CHF)
Common general physical features associated with MVP include the following:
Asthenic body habitusLow body weight or BMIStraight-back syndromeScoliosis or kyphosisPectus excavatumHypermobility of the jointsArm span greater than height (which may be indicative of Marfan syndrome)
The classic auscultatory finding is a mid-to-late systolic click. It may or may not be followed by a high-pitched, mid-to-late systolic murmur at the cardiac apex. These can vary with the following maneuvers:
A Valsalva maneuver or having the patient stand result in an early click, which is close to the first heart sound, and a prolonged murmur The supine position, especially with the legs raised, results in a click later in systole and a shortened murmur
See Clinical Presentation for more detail.
Diagnosis
Findings on echocardiography are as follows:
Classic MVP: The parasternal long-axis view shows > 2 mm superior displacement of the mitral leaflets into the left atrium during systole, with a leaflet thickness of at least 5 mm Nonclassic MVP: Displacement is > 2 mm, with a maximal leaflet thickness of
Other echocardiographic findings that should be considered as criteria are leaflet thickening, redundancy, annular dilatation, and chordal elongation
See Workup for more detail.
Management
For purposes of treatment, patients with MVP can be divided into the following categories:
Asymptomatic patients with minimal diseasePatients with symptoms of autonomic dysfunctionPatients with evidence of progression to severe mitral regurgitationPatients with neurologic findingsPatients with a mid-systolic click and late-systolic mitral regurgitation murmur
Treatment measures for asymptomatic patients with minimal disease
Strong reassurance of the benign prognosisInitial echocardiography for risk stratification; if no clinically significant mitral regurgitation and thin leaflets are observed, clinical examinations and echocardiographic studies can be scheduled every 3-5 years Encouragement to pursue a normal, unrestricted lifestyle, including vigorous exercise
Treatment measures for patients with symptoms of autonomic dysfunction
A trial of beta-blockers for symptomatic reliefAbstinence from stimulants such as caffeine, alcohol, and cigarettesAn ambulatory 24-hour monitor may be useful to detect supraventricular and/or ventricular arrhythmias
Treatment measures for patients with evidence of or progression to severe mitral regurgitation
Close follow-up and early referral for surgical repair, before left ventricular dilatation and systolic dysfunction developSurgery before left ventricular function deteriorates in asymptomatic patients with moderate-to-severe mitral regurgitation and left ventricular enlargement, especially those with atrial fibrillation and/or pulmonary hypertension Treadmill stress test for exercise tolerance if the physician is unsure the patient is asymptomatic
Treatment measures for patients with neurologic findings
After atrial fibrillation and left atrial thrombus are excluded, daily aspirin therapy at a dosage of 80-325 mg/dCessation of smoking and oral contraceptive use to prevent a hypercoagulable stateWarfarin therapy for patients older than 65 years who have atrial fibrillation, especially if they have associated risk factors of a previous stroke or TIA, clinically significant valvular heart disease, hypertension, diabetes, left atrial enlargement, or a history and/or findings of heart failure
Treatment measures for patients with a mid-systolic click and late-systolic mitral regurgitation murmur
Consider antibiotic prophylaxis, including for patients with increased leaflet thickening or redundancyAntibiotic prophylaxis is not recommended for the patient with an isolated mid-to-late systolic click without a murmur, unless the echocardiogram demonstrates significant leaflet redundancy and/or thickness
See Treatment and Medication for more detail.
NextBackground
Mitral valve prolapse (MVP) is the most common valvular abnormality, affecting approximately 2-6% of the population in the United States. MVP usually results in a benign course. However, it occasionally leads to serious complications, including clinically significant mitral regurgitation, infective endocarditis, sudden cardiac death, and cerebrovascular ischemic events. MVP is also the most common cause of isolated mitral regurgitation in the United States, and it is the most common reason for mitral valve surgery.
PreviousNextPathophysiology
Most patients with MVP are asymptomatic, and their natural history is benign. However, when large, floppy valves or ruptured chordae tendinea result in severe mitral regurgitation, mitral valve surgery or repair may be necessary. Myxomatous proliferation is the most common pathologic basis for MVP, and it can lead to myxomatous degeneration of the loose spongiosa and fragmentation of the collagen fibrils. Disruption of the endothelium may predispose patients to infectious endocarditis and thromboembolic complications. However, the vast majority of patients with MVP have only a minor derangement of the mitral valve structure that is usually clinically insignificant.
PreviousNextFrequencyUnited States
MVP is thought to be inherited with increased expression of the gene in female individuals (2:1). The most common form of inheritance is autosomal dominant, but X-linked inheritance has been described.
MVP commonly occurs with heritable connective tissue disorders, including Marfan syndrome, Ehlers-Danlos syndrome, osteogenesis imperfecta, and pseudoxanthoma elasticum. In fact, 90% of patients with Marfan syndrome have MVP due to the increased redundancy of the mitral leaflets and apparatus that occur as a result of myxomatous degeneration.
In the 1970s and 1980s, MVP was overdiagnosed because of the absence of rigorous echocardiographic criteria, with a reported prevalence of 5-15%. Subsequently, Levine et al reported that the 2-dimensional echocardiographic characterizations of prolapse, especially on the parasternal long-axis view, are most specific for the diagnosis of MVP. Use of these criteria prevent overdiagnosis.
Data from the community-based Framingham study demonstrated that MVP syndrome occurred in only 2.4% of the population.
PreviousNextMortality/Morbidity
Most patients with MVP are asymptomatic and have a benign prognosis, with survival rates similar to those of the general population. Nonetheless, high-risk patients (ie, those with moderate-to-severe mitral regurgitation) have increased cardiac morbidity and mortality rates, especially if reduced left ventricular systolic function is present.
See Complications.
Sex
MVP occurs more frequently in young women than in men. The most serious consequences of hemodynamically significant mitral regurgitation occur in men older than 50 years.
Age
MVP has been observed in all ages.
PreviousProceed to Clinical Presentation  Contributor Information and DisclosuresAuthor
Bhavik V Thakkar, MD Medical Director, Internal Medicine Hospitalist, AppleCare Medical Group
Bhavik V Thakkar, MD is a member of the following medical societies: American College of Physicians-American Society of Internal Medicine, American Heart Association, American Medical Association, American Stroke Association, Minnesota Medical Association, and Society for Vascular Medicine and Biology
Disclosure: Nothing to disclose.
Coauthor(s)
Adam E Schussheim, MD Consulting Staff, Department of Internal Medicine, Bridgeport Hospital of the Yale-New Haven Medical Center
Disclosure: Nothing to disclose.
Specialty Editor Board
Justin D Pearlman, MD, ME, PhD, FACC, MA Chief, Division of Cardiology, Director of Cardiology Consultative Service, Director of Cardiology Clinic Service, Director of Cardiology Non-Invasive Laboratory, Director of Cardiology Quality Program KMC, Dartmouth-Hitchcock Medical Center, Dartmouth Medical School
Justin D Pearlman, MD, ME, PhD, FACC, MA is a member of the following medical societies: American College of Cardiology, American College of Physicians, American Federation for Medical Research, International Society for Magnetic Resonance in Medicine, and Radiological Society of North America
Disclosure: Nothing to disclose.
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Medscape Salary Employment
Marschall S Runge, MD, PhD Charles and Anne Sanders Distinguished Professor of Medicine, Chairman, Department of Medicine, Vice Dean for Clinical Affairs, University of North Carolina at Chapel Hill School of Medicine
Marschall S Runge, MD, PhD is a member of the following medical societies: American Association for the Advancement of Science, American College of Cardiology, American College of Physicians-American Society of Internal Medicine, American Federation for Clinical Research, American Federation for Medical Research, American Heart Association, American Physiological Society, American Society for Clinical Investigation, American Society for Investigative Pathology, Association of American Physicians, Association of Professors of Cardiology, Association of Professors of Medicine, Southern Society for Clinical Investigation, and Texas Medical Association
Disclosure: Pfizer Honoraria Speaking and teaching; Merck Honoraria Speaking and teaching; Orthoclinica Diagnostica Consulting fee Consulting
Amer Suleman, MD Private Practice
Amer Suleman, MD is a member of the following medical societies: American College of Physicians, American Heart Association, American Institute of Stress, American Society of Hypertension, Federation of American Societies for Experimental Biology, Royal Society of Medicine, and Society of Cardiac Angiography and Interventions
Disclosure: Nothing to disclose.
Chief Editor
Richard A Lange, MD Professor and Executive Vice Chairman, Department of Medicine, Director, Office of Educational Programs, University of Texas Health Science Center at San Antonio
Richard A Lange, MD is a member of the following medical societies: Alpha Omega Alpha, American College of Cardiology, American Heart Association, and Association of Subspecialty Professors
Disclosure: Nothing to disclose.
Additional Contributors
Alan D Forker, MD Professor of Medicine, University of Missouri at Kansas City School of Medicine; Director, Outpatient Lipid Diabetes Research, MidAmerica Heart Institute of St Luke's Hospital
Disclosure: Nothing to disclose.
References
Izumo M, Shiota M, Kar S, Gurudevan SV, Tolstrup K, Siegel RJ, et al. Comparison of real-time three-dimensional transesophageal echocardiography to two-dimensional transesophageal echocardiography for quantification of mitral valve prolapse in patients with severe mitral regurgitation. Am J Cardiol. Feb 15 2013;111(4):588-94. [Medline].
Perloff JK, Child JS. Mitral valve prolapse. Evolution and refinement of diagnostic techniques. Circulation. Sep 1989;80(3):710-1. [Medline].
Perloff JK, Child JS, Edwards JE. New guidelines for the clinical diagnosis of mitral valve prolapse. Am J Cardiol. May 1 1986;57(13):1124-9. [Medline].
Freed LA, Levy D, Levine RA, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. Jul 1 1999;341(1):1-7. [Medline]. [Full Text].
Levine RA, Triulzi MO, Harrigan P, Weyman AE. The relationship of mitral annular shape to the diagnosis of mitral valve prolapse. Circulation. Apr 1987;75(4):756-67. [Medline].
Alpert MA, Mukerji V, Sabeti M, et al. Mitral valve prolapse, panic disorder, and chest pain. Med Clin North Am. Sep 1991;75(5):1119-33. [Medline].
American College of Cardiology/American Heart Association. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the Manag. J Am Coll Cardiol. 2006;48:1-148:[Medline]. [Full Text].
American College of Cardiology/American Heart Association. ACC/AHA guidelines for the management of patients with valvular heart disease. A report of the American College of Cardiology/American Heart Association. Task Force on Practice Guidelines (Committee on Management of Patients with Valvular Heart Disease. J Am Coll Cardiol. Nov 1998;32(5):1486-588. [Medline]. [Full Text].
Avierinos JF, Gersh BJ, Melton LJ 3rd, Bailey KR, Shub C, Nishimura RA, et al. Natural history of asymptomatic mitral valve prolapse in the community. Circulation. Sep 10 2002;106(11):1355-61. [Medline].
Barnett HJ, Boughner DR, Taylor DW, Cooper PE, Kostuk WJ, Nichol PM. Further evidence relating mitral-valve prolapse to cerebral ischemic events. N Engl J Med. Jan 17 1980;302(3):139-44. [Medline].
Bryhn M, Persson S. The prevalence of mitral valve prolapse in healthy men and women in Sweden. An echocardiographic study. Acta Med Scand. 1984;215(2):157-60. [Medline].
Ciancamerla F, Paglia I, Catuzzo B, Morello M, Mangiardi L. Sudden death in mitral valve prolapse and severe mitral regurgitation. Is chordal rupture an indication to early surgery?. J Cardiovasc Surg (Torino). Apr 2003;44(2):283-6. [Medline].
Clemens JD, Horwitz RI, Jaffe CC, Feinstein AR, Stanton BF. A controlled evaluation of the risk of bacterial endocarditis in persons with mitral-valve prolapse. N Engl J Med. Sep 23 1982;307(13):776-81. [Medline].
Cohn LH, Couper GS, Aranki SF, Rizzo RJ, Kinchla NM, Collins JJ Jr. Long-term results of mitral valve reconstruction for regurgitation of the myxomatous mitral valve. J Thorac Cardiovasc Surg. Jan 1994;107(1):143-50; discussion 150-1. [Medline].
Dajani AS, Taubert KA, Wilson W, et al. Prevention of bacterial endocarditis. Recommendations by the American Heart Association. Circulation. Jul 1 1997;96(1):358-66. [Medline].
David TE, Omran A, Armstrong S, Sun Z, Ivanov J. Long-term results of mitral valve repair for myxomatous disease with and without chordal replacement with expanded polytetrafluoroethylene sutures. J Thorac Cardiovasc Surg. Jun 1998;115(6):1279-85; discussion 1285-6. [Medline].
Davidsen B, Egeblad H, Pietersen A. Thromboembolism in patients with advanced mitral valve prolapse. J Intern Med. Dec 1989;226(6):433-6. [Medline].
Devereux RB. Recent developments in the diagnosis and management of mitral valve prolapse. Curr Opin Cardiol. Mar 1995;10(2):107-16. [Medline].
Devereux RB, Hawkins I, Kramer-Fox R, Lutas EM, Hammond IW, Spitzer MC. Complications of mitral valve prolapse. Disproportionate occurrence in men and older patients. Am J Med. Nov 1986;81(5):751-8. [Medline].
Devereux RB, Kramer-Fox R, Brown WT, Shear MK, Hartman N, Kligfield P. Relation between clinical features of the mitral prolapse syndrome and echocardiographically documented mitral valve prolapse. J Am Coll Cardiol. Oct 1986;8(4):763-72. [Medline].
Devereux RB, Kramer-Fox R, Shear MK, et al. Diagnosis and classification of severity of mitral valve prolapse: methodologic, biologic, and prognostic considerations. Am Heart J. May 1987;113(5):1265-80. [Medline].
Devlin WH, Starling MR. Outcome of valvular heart disease with vasodilator therapy. Compr Ther. 1994;20(10):569-74. [Medline].
Freed LA, Benjamin EJ, Levy D, et al. Mitral valve prolapse in the general population: the benign nature of echocardiographic features in the Framingham Heart Study. J Am Coll Cardiol. Oct 2 2002;40(7):1298-304. [Medline].
Gilon D, Buonanno FS, Joffe MM, et al. Lack of evidence of an association between mitral-valve prolapse and stroke in young patients. N Engl J Med. Jul 1 1999;341(1):8-13. [Medline].
Glesby MJ, Pyeritz RE. Association of mitral valve prolapse and systemic abnormalities of connective tissue. A phenotypic continuum. JAMA. Jul 28 1989;262(4):523-8. [Medline].
Grayburn PA. Vasodilator therapy for chronic aortic and mitral regurgitation. Am J Med Sci. Sep 2000;320(3):202-8. [Medline].
Grigioni F, Enriquez-Sarano M, Ling LH, Bailey KR, Seward JB, Tajik AJ. Sudden death in mitral regurgitation due to flail leaflet. J Am Coll Cardiol. Dec 1999;34(7):2078-85. [Medline].
Kligfield P, Hochreiter C, Kramer H, et al. Complex arrhythmias in mitral regurgitation with and without mitral valve prolapse: contrast to arrhythmias in mitral valve prolapse without mitral regurgitation. Am J Cardiol. Jun 1 1985;55(13 Pt 1):1545-9. [Medline].
Kostuk WJ, Boughner DR, Barnett HJ, Silver MD. Strokes: A complication of mitral-leaflet prolapse?. Lancet. Aug 13 1977;2(8033):313-6. [Medline].
Kramer HM, Kligfield P, Devereux RB, Savage DD, Kramer-Fox R. Arrhythmias in mitral valve prolapse. Effect of selection bias. Arch Intern Med. Dec 1984;144(12):2360-4. [Medline].
Kulan K, Komsuoglu B, Tuncer C, Kulan C. Significance of QT dispersion on ventricular arrhythmias in mitral valve prolapse. Int J Cardiol. Jun 1996;54(3):251-7. [Medline].
Levine RA, Handschumacher MD, Sanfilippo AJ, Hagege AA, Harrigan P, Marshall JE. Three-dimensional echocardiographic reconstruction of the mitral valve, with implications for the diagnosis of mitral valve prolapse. Circulation. Sep 1989;80(3):589-98. [Medline].
Levine RA, Stathogiannis E, Newell JB, et al. Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol. May 1988;11(5):1010-9. [Medline].
Levine RA, Stathogiannis E, Newell JB, et al. Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol. May 1988;11(5):1010-9. [Medline].
Levy D, Savage D. Prevalence and clinical features of mitral valve prolapse. Am Heart J. May 1987;113(5):1281-90. [Medline].
Ling LH, Enriquez-Sarano M, Seward JB, Orszulak TA, Schaff HV, Bailey KR. Early surgery in patients with mitral regurgitation due to flail leaflets: a long-term outcome study. Circulation. Sep 16 1997;96(6):1819-25. [Medline].
MacMahon SW, Hickey AJ, Wilcken DE, Wittes JT, Feneley MP, Hickie JB. Risk of infective endocarditis in mitral valve prolapse with and without precordial systolic murmurs. Am J Cardiol. Jan 1 1987;59(1):105-8. [Medline].
MacMahon SW, Roberts JK, Kramer-Fox R, et al. Mitral valve prolapse and infective endocarditis. Am Heart J. May 1987;113(5):1291-8. [Medline].
Markiewicz W, Stoner J, London E, Hunt SA, Popp RL. Mitral valve prolapse in one hundred presumably healthy young females. Circulation. Mar 1976;53(3):464-73. [Medline].
Marks AR, Choong CY, Sanfilippo AJ, et al. Identification of high-risk and low-risk subgroups of patients with mitral-valve prolapse. N Engl J Med. Apr 20 1989;320(16):1031-6. [Medline].
MartÃnez-Rubio A, Schwammenthal Y, Schwammenthal E, Block M, Reinhardt L, Garcia-Alberola A. Patients with valvular heart disease presenting with sustained ventricular tachyarrhythmias or syncope: results of programmed ventricular stimulation and long-term follow-up. Circulation. Jul 15 1997;96(2):500-8. [Medline].
Mylonakis E, Calderwood SB. Infective endocarditis in adults. N Engl J Med. Nov 1 2001;345(18):1318-30. [Medline].
Nidorf SM, Weyman AE, Hennessey R. The relationship between mitral valve morphology and prognosis in patients with mitral valve prolapse: a prospective echocardiographic study of 568 patients [abstr]. J Am Soc Echocardiogr. 1993;6:S8.
Nishimura RA, McGoon MD. Perspectives on mitral-valve prolapse. N Engl J Med. Jul 1 1999;341(1):48-50. [Medline].
Nishimura RA, McGoon MD, Shub C, Miller FA Jr, Ilstrup DM, Tajik AJ. Echocardiographically documented mitral-valve prolapse. Long-term follow-up of 237 patients. N Engl J Med. Nov 21 1985;313(21):1305-9. [Medline].
Olson LJ, Subramanian R, Ackermann DM, Orszulak TA, Edwards WD. Surgical pathology of the mitral valve: a study of 712 cases spanning 21 years. Mayo Clin Proc. Jan 1987;62(1):22-34. [Medline].
Orencia AJ, Petty GW, Khandheria BK, Annegers JF, Ballard DJ, Sicks JD. Risk of stroke with mitral valve prolapse in population-based cohort study. Stroke. Jan 1995;26(1):7-13. [Medline].
Petty GW, Orencia AJ, Khandheria BK, Whisnant JP. A population-based study of stroke in the setting of mitral valve prolapse: risk factors and infarct subtype classification. Mayo Clin Proc. Jul 1994;69(7):632-4. [Medline].
Procacci PM, Savran SV, Schreiter SL, Bryson AL. Prevalence of clinical mitral-valve prolapse in 1169 young women. N Engl J Med. May 13 1976;294(20):1086-8. [Medline].
Sandok BA, Giuliani ER. Cerebral ischemic events in patients with mitral valve prolapse. Stroke. Jul-Aug 1982;13(4):448-50. [Medline].
Savage DD, Devereux RB, Garrison RJ, et al. Mitral valve prolapse in the general population. 2. Clinical features: the Framingham Study. Am Heart J. Sep 1983;106(3):577-81. [Medline].
Savage DD, Garrison RJ, Devereux RB, Castelli WP, Anderson SJ, Levy D. Mitral valve prolapse in the general population. 1. Epidemiologic features: the Framingham Study. Am Heart J. Sep 1983;106(3):571-6. [Medline].
Savage DD, Levy D, Garrison RJ, Castelli WP, Kligfield P, Devereux RB. Mitral valve prolapse in the general population. 3. Dysrhythmias: the Framingham Study. Am Heart J. Sep 1983;106(3):582-6. [Medline].
Scharf RE, Hennerici M, Bluschke V, Lueck J, Kladetzky RG. Cerebral ischemia in young patients: it is associated with mitral valve prolapse and abnormal platelet activity in vivo?. Stroke. Jul-Aug 1982;13(4):454-8. [Medline].
Tieleman RG, Crijns HJ, Wiesfeld AC, Posma J, Hamer HP, Lie KI. Increased dispersion of refractoriness in the absence of QT prolongation in patients with mitral valve prolapse and ventricular arrhythmias. Br Heart J. Jan 1995;73(1):37-40. [Medline].
Tse HF, Lau CP, Cheng G. Relation between mitral regurgitation and platelet activation. J Am Coll Cardiol. Dec 1997;30(7):1813-8. [Medline].
Ulgen MS, Biyik I, Karadede A, Temamogullari AV, Alan S, Toprak N. Relation between QT dispersion and ventricular arrhythmias in uncomplicated isolated mitral valve prolapse. Jpn Circ J. Dec 1999;63(12):929-33. [Medline].
Warth DC, King ME, Cohen JM, Tesoriero VL, Marcus E, Weyman AE. Prevalence of mitral valve prolapse in normal children. J Am Coll Cardiol. May 1985;5(5):1173-7. [Medline].
Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia, Pa: Saunders; 2005.
Â

 View Table List Read more about Mitral Valve Prolapse on MedscapeRelated Reference Topics
Mitral Valve Prolapse in Emergency Medicine
Pediatric Mitral Valve Prolapse
Mitral Regurgitation
Related News and Articles
Robotic Mitral Valve Surgery
Mild Mitral Regurgitation Common in Young Endurance Athletes
Mitral Valve Surgery
Medscape Reference © 2011 WebMD, LLC, Mitral Valve Prolapse
 Patient positioning when assessing the femoral vein.
Patient positioning when assessing the femoral vein.  Patient positioning when assessing the popliteal vein. PreviousNextTechniqueOverview
Patient positioning when assessing the popliteal vein. PreviousNextTechniqueOverview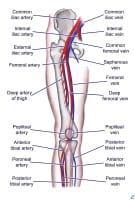 Lower extremity vascular anatomy.
Lower extremity vascular anatomy.  Probe positioning for assessment of the femoral vein.
Probe positioning for assessment of the femoral vein. 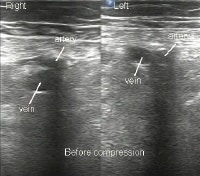 Ultrasonographic image of femoral vessels without compression.
Ultrasonographic image of femoral vessels without compression. 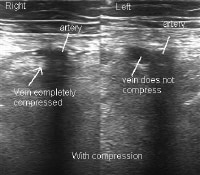 Ultrasonographic image of femoral vessels with compression.
Ultrasonographic image of femoral vessels with compression. 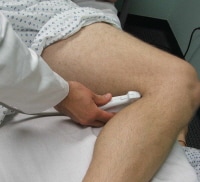 Probe positioning for assessment of the popliteal vein.
Probe positioning for assessment of the popliteal vein. 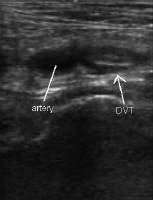 Ultrasonographic image of popliteal vessels with clot.
Ultrasonographic image of popliteal vessels with clot. 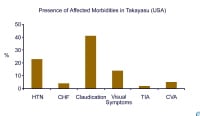 Presence of associated morbidity in Takayasu arteritis in the United States (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
Presence of associated morbidity in Takayasu arteritis in the United States (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr). 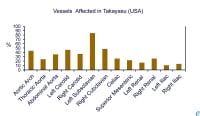 Frequency of vascular involvement (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
Frequency of vascular involvement (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr). 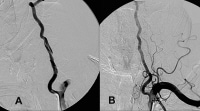 A, Dissection of the left vertebral artery secondary to guidewire injury. B, Complete resolution occurred in 6 months with only aspirin and clopidogrel (Plavix; Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership, Bridgewater, NJ) therapy. NextBackground
A, Dissection of the left vertebral artery secondary to guidewire injury. B, Complete resolution occurred in 6 months with only aspirin and clopidogrel (Plavix; Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership, Bridgewater, NJ) therapy. NextBackground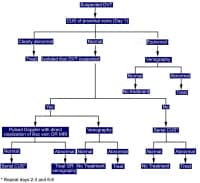 Diagnosis of deep vein thrombosis during pregnancy.
Diagnosis of deep vein thrombosis during pregnancy. 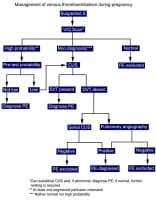 Diagnosis of pulmonary embolism during pregnancy.
Diagnosis of pulmonary embolism during pregnancy. 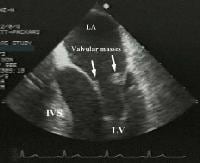 Transesophageal image of a mitral valve with masses characteristic of Libman-Sacks endocarditis.
Transesophageal image of a mitral valve with masses characteristic of Libman-Sacks endocarditis. 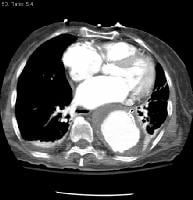 Descending thoracic aortic aneurysm with mural thrombus at the level of the left atrium. NextPathophysiology
Descending thoracic aortic aneurysm with mural thrombus at the level of the left atrium. NextPathophysiology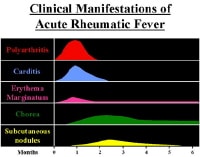 Clinical manifestations and time course. NextPathophysiology
Clinical manifestations and time course. NextPathophysiology










