![]()
Overview of MI Complications
Myocardial infarction (MI) due to coronary artery disease is a leading cause of death in the United States, where more than 1 million people have acute myocardial infarctions (AMIs) each year.[1]
The advent of coronary care units and early reperfusion therapy (lytic or percutaneous coronary intervention) has substantially decreased in-hospital mortality rates and has improved the outcome in survivors of the acute phase of MI.
Complications of MI include arrhythmic, mechanical, and inflammatory (early pericarditis and post-MI syndrome) sequelae, as well as left ventricular mural thrombus (LVMT). In addition to these broad categories, right ventricular (RV) infarction and cardiogenic shock are other possible complications of acute MI. (See the image below.)
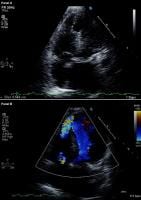
Modified 2-dimensional (top) echocardiogram and color flow Doppler image (bottom). Apical 4-chamber views show a breach in the interventricular septum and free communication between ventricles through a large apical septum ventricular septal defect in a patient who recently had an anterior myocardial infarction.
For other discussions on myocardial infarction, see the overview topics Myocardial Infarction and Right Ventricular Infarction, as well as the articles Imaging of Acute Myocardial Infarcts and Use of Cardiac Markers in the Emergency Department.
NextArrhythmic Complications of MI
About 90% of patients who have an acute myocardial infarction (AMI) develop some form of cardiac arrhythmia during or immediately after the event. In 25% of patients, such rhythm abnormalities manifest within the first 24 hours. In this group of patients, the risk of serious arrhythmias, such as ventricular fibrillation, is greatest in the first hour and declines thereafter. The incidence of arrhythmia is higher with an ST-elevation myocardial infarction (STEMI) and lower with a non–ST-elevation myocardial infarction (NSTEMI).[2]
The clinician must be aware of these arrhythmias, in addition to reperfusion strategies, and must treat those that require intervention to avoid exacerbation of ischemia and subsequent hemodynamic compromise. Most peri-infarct arrhythmias are benign and self-limited. However, those that result in hypotension, increase myocardial oxygen requirements, and/or predispose the patient to develop additional malignant ventricular arrhythmias should be aggressively monitored and treated.
Pathophysiology of arrhythmic complications
AMI is characterized by generalized autonomic dysfunction that results in enhanced automaticity of the myocardium and conduction system. Electrolyte imbalances (eg, hypokalemia and hypomagnesemia) and hypoxia further contribute to the development of cardiac arrhythmia. The damaged myocardium acts as substrate for re-entrant circuits, due to changes in tissue refractoriness.
Enhanced efferent sympathetic activity, increased concentrations of circulating catecholamines, and local release of catecholamines from nerve endings in the heart muscle itself have been proposed to play roles in the development of peri-infarction arrhythmias. Furthermore, transmural infarction can interrupt afferent and efferent limbs of the sympathetic nervous system that innervates myocardium distal to the area of infarction. The net result of this autonomic imbalance is the promotion of arrhythmias.
Classification of peri-infarction arrhythmias
Peri-infarction arrhythmias can be broadly classified into the following categories:
Supraventricular tachyarrhythmias, including sinus tachycardia, premature atrial contractions, paroxysmal supraventricular tachycardia, atrial flutter, and atrial fibrillationAccelerated junctional rhythmsBradyarrhythmias, including sinus bradycardia and junctional bradycardiaAtrioventricular (AV) blocks, including first-degree AV block, second-degree AV block, and third-degree AV blockIntraventricular blocks, including left anterior fascicular block, right bundle branch block (RBBB), and left bundle branch block (LBBB)Ventricular arrhythmias, including premature ventricular contractions (PVCs), accelerated idioventricular rhythm, ventricular tachycardia, and ventricular fibrillationReperfusion arrhythmiasPreviousNextArrhythmic Complications: Supraventricular Tachyarrhythmias
Sinus tachycardia is associated with enhanced sympathetic activity and can result in transient hypertension or hypotension. The elevated heart rate increases myocardial oxygen demand, and a decreased length of diastole compromises coronary flow, worsening myocardial ischemia.
Causes of persistent sinus tachycardia include the following:
Pain AnxietyHeart failureHypovolemiaHypoxiaAnemiaPericarditisPulmonary embolism
In the setting of an AMI, sinus tachycardia must be identified, and appropriate treatment strategies must be devised. Treatment strategies include adequate pain medication, diuresis to manage heart failure, oxygenation, volume repletion for hypovolemia, administration of anti-inflammatory agents to treat pericarditis, and use of beta-blockers and/or nitroglycerin to relieve ischemia.
Premature atrial contractions
Premature atrial contractions often occur before the development of paroxysmal supraventricular tachycardia, atrial flutter, or atrial fibrillation. The usual cause of these extra impulses is atrial distention due to increased left ventricular (LV) diastolic pressure or inflammation associated with pericarditis.
No specific therapy is indicated. However, attention should be given to identifying the underlying disease process, particularly occult heart failure.
Paroxysmal supraventricular tachycardia
The incidence of a paroxysmal supraventricular tachycardia in the setting of an AMI is less than 10%. In the absence of definitive data in the patient with AMI, the consensus is that adenosine can be used when hypotension is not present. In patients without clinically significant LV failure, intravenous diltiazem or a beta-blocker can be used instead. In patients who develop severe heart failure or hypotension, synchronized electrical cardioversion is required.
Atrial flutter
Atrial flutter occurs in less than 5% of patients with AMI. Atrial flutter is usually transient and results from sympathetic overstimulation of the atria.
Treatment strategies for persistent atrial flutter are similar to those for atrial fibrillation, except that ventricular-rate control with drugs is less easily accomplished with atrial flutter than with atrial fibrillation. Therefore, synchronized electrical cardioversion (beginning with 50 J, or the biphasic equivalent) may be needed relatively promptly because of a decrease coronary blood flow and/or hemodynamic compromise. For patients whose atrial flutter is refractory to medical therapy, overdrive atrial pacing may be considered.
Atrial fibrillation
The rate of atrial fibrillation is 10-15% among patients who have AMIs. The onset of atrial fibrillation in the first hours of AMI is usually caused by LV failure, ischemic injury to the atria, or RV infarction. Pericarditis and all conditions leading to elevated left atrial pressure can also lead to atrial fibrillation in association with an AMI. The presence of atrial fibrillation during an AMI is associated with an increased risk of mortality and stroke, particularly in patients who have anterior-wall MIs.
Immediate electrical cardioversion is indicated for the patient in unstable condition, such as one with new or worsening ischemic pain and/or hypotension. Synchronized electrical cardioversion to treat atrial fibrillation begins with 200 J (or the biphasic equivalent). Conscious sedation (preferred) or general anesthesia is advisable prior to cardioversion.
For patients in stable condition, controlling the ventricular response is the immediate objective. If the atrial fibrillation does not respond to cardioversion, IV amiodarone[3] or IV digoxin (in patients with LV dysfunction or heart failure) can be used to achieve ventricular rate control.
For patients who do not develop hypotension, a beta-blocker can be used. For example, metoprolol may be given in 5-mg intravenous boluses every 5-10 min, with a maximum dose of 15 mg. Intravenous diltiazem is an alternative for slowing the ventricular rate, but it should be used with caution in patients with moderate-to-severe heart failure. In patients with new-onset sustained tachycardia (absent before MI), conversion to sinus rhythm should be considered as an option.
Atrial fibrillation and atrial flutter confer an increased risk of thromboembolism (see Deep Venous Thrombosis and Pulmonary Embolism). Therefore, anticoagulation with either unfractionated heparin or low molecular weight heparin (LMWH) should be started if contraindications are absent. It is unclear whether anticoagulation is needed in cases of transient atrial fibrillation and how long after the onset of atrial fibrillation should the anticoagulation be started.
PreviousNextArrhythmic Complications: Accelerated Junctional Rhythm
An accelerated junctional rhythm results from increased automaticity of the junctional tissue that leads to a heart rate of 70-130 bpm. This type of dysrhythmia is most common in patients who develop inferior myocardial infarctions. Treatment is directed at correcting the underlying ischemia.
PreviousNextArrhythmic Complications: BradyarrhythmiasSinus bradycardia
Sinus bradycardia is a common arrhythmia in patients with inferior or posterior acute myocardial infarctions (AMIs). The highest incidence, 40%, is observed in the first 1-2 hours after AMI.
The likely mechanism leading to bradycardia and hypotension is stimulation of cardiac vagal afferent receptors that result in efferent cholinergic stimulation of the heart. In the early phases of an AMI, resultant sinus bradycardia may actually be protective, reducing myocardial oxygen demand. Clinically significant bradycardia that decreases cardiac output and hypotension may result in ventricular arrhythmias and should, therefore, be treated aggressively. Isolated sinus bradycardia is not associated with an increase in the acute mortality risk, and therapy is typically unnecessary when the patient has no adverse signs or symptoms.
When emergency therapy is indicated (eg, in a patient with a sinus rate of
When atropine is ineffective and the patient is symptomatic or hypotensive, transcutaneous or transvenous pacing is indicated (see our main article on External Pacemakers). Denervate, transplanted hearts do not respond to atropine and, therefore, require cardiac pacing.
If these interventions fail, additional pharmacologic intervention may be useful. Examples are dopamine 5-20 mcg/kg/min given intravenously, epinephrine 2-10 mcg/min, and/or dobutamine.
Junctional bradycardia
Junctional bradycardia is a protective AV junctional escape rhythm at a rate of 35-60 bpm in patients who have an inferior MI. This arrhythmia is not usually associated with hemodynamic compromise, and treatment is typically not required.
PreviousNextArrhythmic Complications: AV and Intraventricular BlocksFirst-degree AV block
First-degree AV block is characterized by prolongation of the PR interval to longer than 0.20 seconds. This type of block occurs in approximately 15% of patients who have an acute myocardial infarction (AMI), most commonly an inferior infarction. Almost all patients who develop first-degree AV block have conduction disturbances above the His bundle. In these patients, the progression to complete heart block or ventricular asystole is rare. No specific therapy is indicated unless associated hemodynamic compromise is present.
Calcium channel blockers and beta-blockers may cause or exacerbate a first-degree AV block, but they should be stopped only if hemodynamic impairment or a higher-degree block occurs. For a first-degree AV block associated with sinus bradycardia and hypotension, atropine should be administered. Continued cardiac monitoring is advisable in view of possible progression to higher degrees of block.
Second-degree AV block
Mobitz type I, or Wenckebach, AV block occurs in approximately 10% of patients who have an AMI and accounts for 90% of all patients who have an AMI and a second-degree AV block. A second-degree AV block is associated with a narrow QRS complex and is most commonly associated with an inferior MI. It does not affect the patient's overall prognosis.
A Mobitz type I block does not necessarily require treatment. If the heart rate is inadequate for perfusion, immediate treatment with atropine 0.5-1 mg administered intravenously is indicated. Transcutaneous or temporary transvenous pacing is rarely required.
A Mobitz type II AV block accounts for 10% of all second-degree AV blocks (overall rate of
Mobitz type II AV blocks are associated with a poor prognosis, as the mortality rate associated with their progression to a complete heart block is approximately 80%. Therefore, this type of second-degree AV block should be immediately treated with transcutaneous pacing or atropine. Atropine helps in about 50% of cases, but it occasionally worsens the block with an increased heart rate. A temporary transvenous pacemaker, and possibly a permanent demand pacemaker, must ultimately be placed.
Third-degree AV block
A third-degree AV block (ie, a complete heart block), occurs in 5-15% of patients who have an AMI and may occur with anterior or inferior infarctions. In patients with inferior infarctions, this type of block usually develops gradually, progressing from first-degree or a type I second-degree block. In most patients, the level of the block is supranodal or intranodal, and the escape rhythm is usually stable with a narrow QRS and rates exceeding 40 bpm. In 30% of patients, the block is below the His bundle, where it results in an escape rhythm with a rate slower than 40 bpm and a wide QRS complex.
Complete heart block in patients with an inferior MI usually responds to atropine. In most patients, it resolves within a few days without the need for a temporary or permanent pacemaker. The mortality rate for patients with inferior MI who develop complete heart block is approximately 15% unless a coexisting RV infarction is present, in which case the mortality rate is higher.
Immediate treatment with atropine is indicated for patients with third-degree AV blocks. As with therapy for a Mobitz type II block, this treatment may not help and may sometimes worsen the block. Temporary transcutaneous or transvenous pacing is indicated for symptomatic patients whose condition is unresponsive to atropine. Permanent pacing should be considered in patients with persistent symptomatic bradycardia that remains unresolved with lysis or percutaneous coronary intervention.
In patients with an anterior MI, an intraventricular block or a Mobitz type II AV block usually precedes a third-degree AV block. The third-degree block occurs suddenly and is associated with a high mortality rate. The Cardiac Arrhythmias and Risk Stratification After Myocardial Infarction (CARISMA) trial monitored patients with acute myocardial infarction and reduced left ventricular ejection fraction and found that high-degree atrioventricular block was the most powerful predictor of cardiac death.[4] Patients with these blocks typically have unstable escape rhythms with wide QRS complexes and at rates of less than 40 bpm.
Immediate treatment with atropine and/or transcutaneous pacing is indicated. This is followed by temporary transvenous pacing. Patients with an anterior MI who develop a third-degree AV block and who survive to hospitalization often receive a permanent pacemaker.
Intraventricular blocks
Conduction from the His bundle is transmitted through 3 fascicles: the anterior division of the left bundle, the posterior division of the left bundle, and the right bundle. An abnormality of electrical conduction in 1 or more of these fascicles is noted in about 15% of patients with AMI. Isolated left anterior fascicular block (LAFB) occurs in 3-5% of patients with AMI; progression to complete AV block is uncommon. Isolated left posterior fascicular block occurs in only 1-2% of patients who have an AMI. The blood supply of the posterior fascicle is larger than that of the anterior fascicle; therefore, a block here is associated with a relatively large infarct and high mortality rate.
The right bundle branch receives its dominant blood supply from the left anterior descending (LAD) artery. Therefore, a new RBBB, which is seen in approximately 2% of patients with AMI, suggests a large infarct territory. However, progression to complete heart block is uncommon. In patients who develop an anterior MI and a new RBBB, the substantial risk for death is mostly from cardiogenic shock, which is presumably due to the large size of the myocardial infarct.
The combination of RBBB with an LAFB is known as bifascicular block and commonly occurs with occlusion of the proximal LAD coronary artery. The risk of developing complete AV block is heightened, but complete block is still uncommon. Mortality is mostly related to the amount of muscle loss. Bifascicular block in the presence of first-degree AV block is called a trifascicular block. In 40% of patients, a trifascicular block progresses to a complete heart block.
PreviousNextArrhythmic Complications: Ventricular ArrhythmiasPremature ventricular contractions
In the past, frequent premature ventricular contractions (PVCs) were considered to represent warning arrhythmias and indicators of impending malignant ventricular arrhythmias. However, presumed warning arrhythmias are frequently observed in patients who have an acute myocardial infarction (AMI) and who never develop ventricular fibrillation. On the converse, primary ventricular fibrillation often occurs without antecedent premature ventricular ectopy.
For these reasons, prophylactic suppression of PVCs with antiarrhythmic drugs, such as lidocaine, is no longer recommended. Prophylaxis has been associated with an increased risk of fatal bradycardia or asystole because of the suppression of escape pacemakers.
Given this evidence, most clinicians pursue a conservative course when PVCs are observed in a patient with an AMI, and they do not routinely administer prophylactic antiarrhythmics. Instead, attention should be directed toward correcting any electrolytic or metabolic abnormalities, plus identifying and treating recurrent ischemia.
Accelerated idioventricular rhythm
An accelerated idioventricular rhythm is seen in as many as 20% of patients who have an AMI. This pattern is defined as a ventricular rhythm characterized by a wide QRS complex with a regular escape rate faster than the atrial rate, but less than 100 bpm. AV dissociation is frequent. Slow, nonconducted P waves are seen; these are unrelated to the fast, wide QRS rhythm.
Most episodes are short and terminate spontaneously. They occur with equal frequency in anterior and inferior infarctions. The mechanism might involve (1) the sinoatrial node or the AV node, which may sustain structural damage and depress nodal automaticity, and/or (2) an abnormal ectopic focus in the ventricle that takes over as the dominant pacemaker.
The presence of accelerated idioventricular rhythm does not affect the patient's prognosis; no definitive evidence has shown that an untreated occurrence increases the incidence of ventricular fibrillation or death. This rhythm occurs somewhat more frequently in patients who develop early reperfusion than in others; however, it is neither sensitive nor specific as a marker of reperfusion.
Temporary pacing is not indicated unless the rhythm is sustained and results in hypotension or ischemic symptoms. An accelerated idioventricular rhythm represents an appropriate escape rhythm. Suppression of this escape rhythm with an antiarrhythmic drug can result in clinically significant bradycardia or asystole. Therefore, an accelerated idioventricular rhythm should be left untreated.
Nonsustained ventricular tachycardia
Nonsustained ventricular tachycardia is defined as 3 or more consecutive ventricular ectopic beats at a rate of greater than 100 bpm and lasting less than 30 seconds. In patients who experience multiple runs of nonsustained ventricular tachycardia, the risk for sudden hemodynamic collapse may be substantial.
Nonetheless, nonsustained ventricular tachycardia in the immediate peri-infarction period does not appear to be associated with an increased mortality risk, and no evidence suggests that antiarrhythmic treatment offers a morbidity or mortality benefit. However, nonsustained ventricular tachycardia occurring more than 48 hours after infarction in patients with LV systolic dysfunction (LV ejection fraction
Multiple episodes of nonsustained ventricular tachycardia require intensified monitoring and attention to electrolyte imbalances. Serum potassium levels should be maintained above 4.5 mEq/L, and serum magnesium levels should be kept above 2.0 mEq/L. Ongoing ischemia should aggressively be sought and corrected if found.
Sustained ventricular tachycardia
Sustained ventricular tachycardia is defined as 3 or more consecutive ventricular ectopic beats at a rate greater than 100 bpm and lasting longer than 30 seconds or causing hemodynamic compromise that requires intervention. Monomorphic ventricular tachycardia is most likely to be caused by a myocardial scar, whereas polymorphic ventricular tachycardia may be most responsive to measures directed against ischemia. Sustained polymorphic ventricular tachycardia after an AMI is associated with a hospital mortality rate of 20%.
Emergency treatment of sustained ventricular tachycardia is mandatory because of its hemodynamic effects and because it frequently deteriorates into ventricular fibrillation. Rapid polymorphic ventricular tachycardia (rate >150 bpm) associated with hemodynamic instability should be treated with immediate direct-current unsynchronized cardioversion of 200 J (or biphasic energy equivalent). Monomorphic ventricular tachycardia should be treated with a synchronized discharge of 100 J (or biphasic energy equivalent).
If sustained ventricular tachycardia is well tolerated, antiarrhythmic therapy with amiodarone (drug of choice) or procainamide may be attempted before electrical cardioversion. Precipitating causes, such as electrolyte abnormalities, acid-base disturbances, hypoxia, or medication, should be sought and corrected. For persistent or recurrent ventricular tachycardia, overdrive pacing may be effective in electrically converting the patient's rhythm to a sinus rhythm.
Ventricular fibrillation
The incidence of primary ventricular fibrillation is greatest in the first hour after the onset of infarct (4.5%) and declines rapidly thereafter. Approximately 60% of episodes occur within 4 hours, and 80% occur within 12 hours.
Secondary or late ventricular fibrillation occurring more than 48 hours after an MI is usually associated with pump failure and cardiogenic shock. Factors associated with an increased risk of secondary ventricular fibrillation are a large infarct, an intraventricular conduction delay, and an anteroseptal AMI. Secondary ventricular fibrillation in conjunction with cardiogenic shock is associated with an in-hospital mortality rate of 40-60%.
Treatment for ventricular fibrillation is unsynchronized electrical countershock with at least 200-300 J (or biphasic energy equivalent) administered as rapidly as possible. Each minute after the onset of uncorrected ventricular fibrillation is associated a 10% decrease in the likelihood of survival. Restoration of synchronous cardiac electrical activity without the return of effective contraction (ie, electromechanical dissociation, or pulseless electrical activity) is generally due to extensive myocardial ischemia and/or necrosis or cardiac rupture.
Antiarrhythmics, such as intravenous amiodarone and lidocaine, facilitate successful electrical defibrillation and help prevent recurrent or refractory episodes. After ventricular fibrillation is successfully converted, antiarrhythmic therapy is generally continued as a constant intravenous infusion for 12-24 hours.
Prophylactic lidocaine reduces the incidence of ventricular fibrillation, but it is not used because it seems to be associated with an excessive mortality risk owing to bradycardic and asystolic events[5] . On the other hand, early use of beta-blockers in patients with AMI reduces the incidence of ventricular fibrillation as well as death[6] .
PreviousNextArrhythmic Complications: Reperfusion Arrhythmias
In the past, the sudden onset of rhythm disturbances after thrombolytic therapy in patients with AMI was believed to be a marker of successful coronary reperfusion. However, a high incidence of identical rhythm disturbances is observed in patients with AMI in whom coronary reperfusion is unsuccessful. Therefore, these so-called reperfusion arrhythmias are neither sensitive nor specific for reperfusion and should be treated as discussed under Accelerated Idioventricular Rhythm in the Arrhythmic Complications: Ventricular Arrhythmias section above.
PreviousNextMechanical Complications of MI
The 3 major mechanical complications of AMI are ventricular free wall rupture (VFWR), ventricular septal rupture (VSR), and papillary muscle rupture with severe mitral regurgitation (MR). Each of these complications can result in cardiogenic shock. Clinical issues related to these mechanical problems are discussed below. (See also Myocardial Rupture.)
Overview of ventricular free wall rupture
VFWR is the most serious complication of AMI. VFWR is usually associated with large transmural infarctions and antecedent infarct expansion. It is the most common cause of death, second only to LV failure, and it accounts for 15-30% of the deaths associated with AMI. Incontrovertibly the most catastrophic of mechanical complications, VFWR leads to acute hemopericardium and death from cardiac tamponade.
The overall incidence of VFWR ranges from 0.8-6.2%. The incidence of this complication has declined over the years with better 24 hour systolic blood pressure control; increased use of reperfusion therapy, beta blockers, and ACE inhibitors; and decreased use of heparin[7] .
Data from the National Registry of Myocardial Infarction (NRMI) showed an elevated incidence of in-hospital mortality among patients who received thrombolytic therapy (12.1%) than among patients who did not (6.1%).[8] In the Thrombolysis in Myocardial Infarction Phase II (TIMI II) trial, 16% of patients died from cardiac rupture within 18 hours of therapy.[9] Patients who underwent percutaneous transluminal coronary angioplasty (PTCA) had an incidence of free wall rupture lower than that of patients receiving thrombolytic therapy.
Risk factors for VFWR include advanced age greater than 70 years, female sex, no previous MIs, Q waves on ECG, hypertension during the initial phase of STEMI, corticosteroid or NSAID use, and fibrinolytic therapy more than 14 hours after STEMI onset. Patients with a history of angina pectoris, previous AMI, multivessel coronary disease, and chronic heart failure are less likely than others to develop VFWR of the LV because they develop collaterals and ischemic preconditioning.[8, 10, 11]
Clinical presentation of VFWR
VFWRs are dramatic; they present acutely or occasionally subacutely as pseudoaneurysms; and they most often involve the anterior or lateral wall of the LV. Most VFWRs occur within the first week after AMI.
Becker et al classified the following 3 types of VFWRs[12] :
Type I - an abrupt slitlike tear that is frequently associated with anterior infarcts and that occurs early (within 24 h)Type II - an erosion of infarcted myocardium at the border between the infarcted and viable myocardiumType III - an early aneurysm formation correlated with older and severely expanded infarcts
Type III usually occurs later than type I or type II ruptures. Thrombolytic therapy accelerates the occurrence of cardiac rupture in Becker type I and type II VFWRs. In severely expanded infarctions (type III), thrombolytic therapy decreases the incidence of cardiac rupture.
A pseudoaneurysm is formed when adjacent pericardium and hematoma seals off a myocardial rupture or perforation. The wall of a pseudoaneurysm is most often visualized as an aneurysmal outpouching that communicates with the LV cavity by means of a narrow neck. This wall is composed of pericardium and organized thrombus and/or hematoma. It is devoid of myocardial elements, whereas a true aneurysm has all the elements of the original myocardial wall and a relatively wide base. The pseudoaneurysm may vary in size and is at high risk of rupturing.
Clinical presentations of VFWR vary depending on the acuity, location, and size of the rupture. Patients with acute VFWR present with severe chest pain, abrupt electromechanical dissociation or asystole, hemodynamic collapse, and possibly death. In about one third of the patients, the course is subacute, and they present with symptoms such as syncope, hypotension, shock, arrhythmia, and prolonged and recurrent chest pain.
Diagnosis of VFWR
Early diagnosis of VFWRs and intervention are critical to patient survival. A high index of suspicion is required when patients with AMI present with severe chest pain, shock or arrhythmias, and abrupt development of electromechanical dissociation. ECG signs of impending VFWR have limited specificity but include sinus tachycardia, intraventricular conduction defect, and persistent or recurrent ST-segment elevation.
Echocardiography is the diagnostic tool of choice. The key diagnostic finding is a moderate-to-large pericardial effusion with clinical and echocardiographic signs of impending pericardial tamponade. In patients with cardiac tamponade and electromechanical dissociation, moderate-to-severe pericardial effusion increases the mortality risk. Those patients without initial cardiac tamponade, while at a lower rate of mortality, should still be followed, as late rupture may still occur.[13] The absence of pericardial effusion on echocardiography has high negative predictive value. If the ability to obtain transthoracic echocardiograms is limited in patients receiving mechanical ventilation, transesophageal echocardiography can assist in confirming VFWR.
MRI provides superior image quality and permits identification of the site and anatomy of a ventricular pseudoaneurysm (ie, ruptured LV restrained by the pericardium with enclosed clot). However, MRI is of limited use in the acute setting because of the time involved and nonportability of imaging units.
Treatment of VFWR
The most important prevention strategy is early reperfusion therapy, with percutaneous coronary intervention (PCI) being the preferred modality. Fibrinolytic therapy is associated with overall decreased risk of VFWR; however, its use more than 14 hours after STEMI onset can increase the risk of early rupture.[14, 15]
The standard treatment for VFWR is emergency surgical repair after hemodynamic stability is achieved. Patients may first need intravenous fluids, inotropic agents, and emergency pericardiocentesis.
Pifarré and associates recommended the deployment of an intra-aortic balloon pump to decrease systolic afterload and improve diastolic myocardial perfusion.[16]
Several surgical techniques have been applied, including infarctectomy, adhering with biologic glue patches made of polyethylene terephthalate polyester fiber (Dacron; DuPont, Wilmington, DE) or polytetrafluoroethylene fluoropolymer resin (Teflon; DuPont); and use of pledgeted sutures without infarctectomy.
The mortality rate is significantly high and largely depends on the patient's preoperative hemodynamic status. Early diagnosis, rapid institution of the measures described above to achieve hemodynamic stability, and prompt surgical repair can improve survival rates. A follow-up to the Acorn randomized trial demonstrated long-term improvement in left ventricular structure and function after mitral valve surgery for as long as 5 years. These data provide evidence supporting mitral valve repair in combination with the Acorn CorCap device for patients with nonischemic heart failure with severe left ventricular dysfunction who have been medically optimized yet remain symptomatic with significant mitral regurgitation.[17]
Overview of ventricular septal rupture
VSR is an infrequent but life-threatening complication of AMI. Despite optimal medical and surgical treatment, patients with VSR have a high in-hospital mortality rate. During the prethrombolytic era, VSRs occurred in 1-3% of individuals with MIs. The incidence declined with thrombolytic therapy (to 0.2-0.34%) because of improvements in reperfusion and myocardial salvage. The bimodal distribution of VSR is characterized by a high incidence in the first 24 hours, with another peak on days 3-5 and rarely more than 2 weeks after AMI.
In patients receiving thrombolytics, the median time from the onset of symptoms of AMI to septal rupture was 1 day in the Global Utilization of Streptokinase and TPA [tissue plasminogen activator] for Occluded Coronary Arteries (GUSTO-I) trial[18] and 16 hours in the Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock? (SHOCK) trial.[19]
Risk factors for septal rupture include advanced age (>65 y), female sex, single-vessel disease, extensive MI, and poor septal collateral circulation.[20, 21] Before the advent of thrombolytics, hypertension and absence of a history of angina were risk factors for VSR. Extensive infarct size and RV involvement are other known risk factors for septal rupture.
In patients with AMI without reperfusion, coagulation necrosis develops within 3-5 days after infarction. Neutrophils migrate to the necrotic zone and undergo apoptosis, release lytic enzymes, and hasten the disintegration of necrotic myocardium. Some patients have infarcts with large intramural hematomas, which dissect into the tissue and result in early septal rupture. The size of the septal rupture ranges from a few millimeters to several centimeters.
VSR is categorized as simple or complex depending on its length, course, and location. In simple septal rupture, the perforation is at the same level on both sides of the septum, and a direct through-and-through communication is present across the septum. A complex septal rupture is characterized by extensive hemorrhage with irregular, serpiginous tracts in the necrotic tissue.
Septal ruptures are most common in patients with large anterior MIs due to occlusion of the LAD artery causing extensive septal infarcts. These infarcts are associated with ST-segment elevations and Q waves in inferior leads (II, III, aVF) and these ECG changes are therefore more commonly seen in septal ruptures.[22] These ruptures are generally apical and simple.
Septal ruptures in patients with inferior MI occur relatively infrequently. These ruptures involve the basal inferoposterior septum and are often complex.
Clinical presentation of VSR
Symptoms of VSR complicating AMI include chest pain, shortness of breath, hypotension, biventricular failure, and shock within hours to days. Patients often present with a new, loud, and harsh holosystolic murmur. This murmur is loudest along the lower left sternal border and is associated with a palpable parasternal systolic thrill. RV and LV S3 gallops are common.
In patients with cardiogenic shock complicating septal rupture, the murmur and thrill may be difficult to identify. In contrast, patients with acute MR often have a soft systolic murmur at the apex without a thrill.
Diagnosis of VSR
Echocardiography with color flow Doppler imaging is the diagnostic tool of choice for identifying a VSR. (See the image below.) Its sensitivity and specificity have been reported to be as high as 100%. In addition, it can be used for the following:
Define the site and size of septal ruptureAssess the LV and RV functionEstimate the RV systolic pressureQuantify the left-to-right shunt
Cardiac catheterization is usually required to confirm the diagnosis, quantitate the degree of left-to-right shunt, differentiate VSR from other conditions (eg, mitral regurgitation), plus visualize the coronary arteries.
Modified 2-dimensional (top) echocardiogram and color flow Doppler image (bottom). Apical 4-chamber views show a breach in the interventricular septum and free communication between ventricles through a large apical septum ventricular septal defect in a patient who recently had an anterior myocardial infarction.
In patients with VSR, right-heart catheterization shows a step-up in oxygen saturation from the right atrium to the RV; in contrast, no step-up in oxygen saturation occurs among patients with MR. The presence of large V waves in the pulmonary capillary wedge tracing supports the diagnosis of severe acute MR.
Left ventriculography can also be used to identify the site of ventricular rupture (see Cardiac Catheterization [Left Heart]). However, this study is usually unnecessary after a good-quality echocardiographic and Doppler examination is conducted.
Treatment of VSR
The key to management of VSR is prompt diagnosis and an aggressive approach to hemodynamic stabilization, angiography, and surgery. The optimal approach includes hemodynamic stabilization with the administration of oxygen and mechanical support with use of an intra-aortic balloon pump, as well as the administration of vasodilators (to reduce afterload and thus LV pressure and the left-to-right shunt), diuretics, and inotropic agents.
Cardiac catheterization is needed to define the coronary anatomy; this is followed by urgent surgical repair.
Medical therapy is intended only for temporary stabilization before surgery, as most patients' conditions deteriorate rapidly and they die in the absence of surgical intervention. In the GUSTO-I trial, the 30-day mortality rate was lower in patients with VSR who underwent surgical repair than in patients treated medically (47% vs 94%), as was the 1-year mortality rate (53% vs 97%).[18] Lemery et al reported a 30-day survival rate of 24% in patients treated medically compared with 47% in those treated surgically.[23]
Current guidelines of the American College of Cardiology/American Heart Association for the treatment of patients with septal rupture complicating AMI highlight urgent surgical intervention, regardless of their clinical status. Surgical management of septal rupture includes the following elements:
Prompt establishment of hypothermic cardiopulmonary bypassAn approach to the septal rupture through the infarct area and the excision of all necrotic, friable margins of the septum and ventricular walls to avoid postoperative hemorrhage, residual septal defect, or both Reconstruction of the septum and ventricular walls by using prosthetic material and preservation of the geometric configuration of the ventricles and heart function
Percutaneous closure of septal rupture is a relatively new approach, one used in select patients as an alternative to surgical repair or for the acute stabilization of critically ill patients. However, percutaneous closure is currently unavailable in many institutions, and no long-term outcome data are available.
Several studies failed to show a relationship between perioperative mortality and concomitant coronary revascularization (coronary artery bypass grafting). Patients with cardiogenic shock due to septal rupture have the poorest outcome. In the SHOCK trial, the in-hospital mortality rate was higher in patients with cardiogenic shock due to septal rupture (87.3%) than in patients with cardiogenic shock from all other causes (59.2% with pure LV failure and 55.1% with acute MR).[19, 24]
In patients who survive surgical repair, the rate of recurrent or residual septal defect is reported to be about 28%, and the associated mortality rate is high.
Repeat surgical intervention is indicated in patients who have clinical heart failure or a pulmonary-systemic fraction greater than 2.
Overview of acute mitral regurgitation
MR is a common complication of AMI that results from local and global LV remodeling and that is an independent predictor of heart failure and death. MR typically occurs 7-10 days after an AMI, though this onset may vary according to the mechanism of MR. Papillary muscle rupture resulting in MR occurs within 1-14 days (median, 1 d).
Mild-to-moderate MR is often clinically silent and detected on Doppler echocardiography performed during the early phase of AMI. In such cases, MR rarely causes hemodynamic compromise.
Speckle tracking and 3-dimensional echocardiography proved to be important imaging tools in assessing reverse LV remodeling after degenerative mitral valve regurgitation surgery. Subtle regional preoperative changes in diastolic function of the septal and lateral wall could be preoperatively identified, aiding in optimizing the referral timing and recognizing potential culprits as indicators of disease recurrence after mitral repair.[25]
Severe acute MR that results from the rupture of papillary muscles or chordae tendineae results in abrupt hemodynamic deterioration with cardiogenic shock. Rapid diagnosis, hemodynamic stabilization, and prompt surgical intervention are needed because acute severe MR is associated with a high mortality rate.
The reported incidence of MR may vary because of several factors, including the diagnostic methods used, the presence or absence of heart failure, the degree of MR reported, the type of therapy rendered, and the time from infarct onset to testing.
During the GUSTO-I trial, the incidence of MR in patients receiving thrombolytic therapy was 1.73%.[18] The SHOCK trial, which included MI patients presenting with cardiogenic shock, noted a 39.1% incidence of moderate to severe MR.[26] Kinn et al reported that reperfusion with angioplasty resulted in an 82% decrease in the rate of acute MR, as compared with thrombolytic therapy (0.31% vs 1.73%).[27]
Risk factors for MR are advanced age, female sex, large infarct, previous AMI, recurrent ischemia, multivessel coronary artery disease, and heart failure.
Several mechanisms can cause MR after AMI. Rupture of the papillary muscle is the most commonly reported mechanism.
Such rupture occurs in 1% of patients with AMI and frequently involves the posteromedial papillary muscle rather than the anterolateral papillary muscle, as the former has a single blood supply versus the dual supply for the latter. Papillary muscle rupture may lead to flailing or prolapse of the leaflets, resulting in severe MR. Papillary muscle dysfunction due to scarring or recurrent ischemia may also lead to MR in the subacute and chronic phases after MI; this condition can resolve spontaneously.
Large posterior infarctions produce acute MR due to asymmetric annular dilation and altered function and geometry of the papillary muscle.
Clinical presentation of MR
Patients with functional mild or moderate MR are often asymptomatic. The severity of symptoms varies depending on ventricular function. Clinical features of acute severe MR include shortness of breath, fatigue, a new apical holosystolic murmur, flash pulmonary edema, and shock.
The new systolic murmur may be only early-to-mid systolic, not holosystolic. It may be soft or even absent because of the abrupt rise in left atrial pressure, which lessens the pressure gradient between the left atrium and the LV, as compared with chronic MR. The murmur is best heard at the apex rather than the lower left sternal border, and it is uncommonly associated with a thrill. S3 and S4 gallops are expected.
Diagnosis of MR
The clinician cannot rely on a new holosystolic murmur to diagnose MR or assess its severity because of the variable hemodynamic status. In a patient with AMI who presents with a new apical systolic murmur, acute pulmonary edema, and cardiogenic shock, a high index of clinical suspicion for severe MR is the key to diagnosis.
Chest radiography may show evidence of pulmonary edema in the acute setting without clinically significant cardiac enlargement.
Echocardiography with color flow Doppler imaging is the standard diagnostic tool for detecting MR. Transthoracic echocardiography is the preferred initial screening tool, but transesophageal echocardiography is invaluable in defining the severity and exact mechanism of acute MR, especially when suspicion for papillary muscle rupture is high. Cardiac catheterization should be performed in all patients to determine the extent and severity of coronary artery disease.
Treatment of MR
Determination of hemodynamic stability, elucidation of the exact mechanism of acute MR, and expedient therapy are all necessary for a favorable outcome. Medical management includes afterload reduction with the use of diuretics, sodium nitroprusside, and nitrates in patients who are not hypotensive.
In patients who have hemodynamic compromise, intra-aortic balloon counterpulsation should be deployed rapidly. This intervention usually substantially reduces afterload and regurgitant volume, improving cardiac output in preparation for surgical repair. Without surgical repair, medical therapy alone in patients with papillary muscle rupture results in inadequate hemodynamic improvement and a poor short-term prognosis.
Emergency surgical intervention is the treatment of choice for papillary muscle rupture. Surgical approaches may include mitral valve repair or replacement. In the absence of papillary muscle necrosis, mitral valve repair improves the survival rate more than mitral valve replacement does. This difference is because the subvalvular apparatus is usually preserved. Mitral valve repair also eliminates complications related to malfunction of the prosthesis.
In patients with extensive necrosis of papillary muscle and/or ventricular free wall, mitral valve replacement is the preferred modality. Coronary artery bypass grafting (CABG) performed at the time of surgery was shown in one study to improve short- and long-term survival.[28]
The only situation in which emergency surgery can safely be avoided is in the case of intermittent MR due to recurrent ischemia. In these patients, successful myocardial revascularization may be effective. This procedure is accomplished by means of either angioplasty or coronary artery bypass grafting.
PreviousNextLeft Ventricular AneurysmOverview of LVA
Left ventricular aneurysm (LVA) is defined as a localized area of myocardium with abnormal outward bulging and deformation during both systole and diastole. The rate of LVAs after AMI is approximately 3-15%. Risk factors for LVA after AMI include female sex, total occlusion of the LAD artery, single-vessel disease, and absence of previous angina.
More than 80% of LVAs affect the anterolateral wall; these are usually associated with total occlusion of the LAD. The posterior and inferior walls are less commonly affected. LVAs generally range from 1-8 cm. Histologically, LVAs are composed of fibrous scar that is notably thinned. This scar is clearly delineated from the adjacent ventricular muscle on microscopic examination.
A history of MI and third or fourth heart sounds are common findings from the patient's history and physical examination.
Diagnosis of LVA
The chest radiograph may reveal an enlarged cardiac silhouette.
Electrocardiography is characterized by ST elevation that persists several weeks after AMI and that appears in the same leads as those showing the acute infarct. Echocardiography is 93% sensitive and 94% specific for detection of LVA (see the image below), but cardiac catheterization remains the standard for establishing the diagnosis.
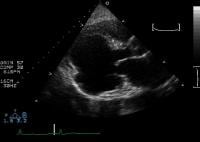
Parasternal long-axis view of the left ventricle demonstrates a large inferobasal aneurysm. Note the wide neck and base of the aneurysm. Treatment of LVA
Patients with small or clinically insignificant aneurysms can be treated conservatively with close follow-up. Medical therapy generally consists of the use of angiotensin-converting enzyme (ACE) inhibitors, which reduce afterload, infarct extension, and LV remodeling. Anticoagulation is required when patients have severe LV dysfunction and/or thrombus in the LV or aneurysm.
Surgical resection of the LVA is indicated if severe heart failure, ventricular tachyarrhythmias refractory to medical treatment, or recurrent thromboembolism is present.
PreviousNextMiscellaneous ComplicationsLeft ventricular mural thrombus
LVMT is a well-known complication of AMI and frequently develops after anterior infarcts of the LV wall. The incidence of LVMT as a complication of AMI ranges from 20-40% and may reach 60% in patients with large anterior-wall AMIs who are not treated with anticoagulant therapy. LVMT is associated with a high risk of systemic embolization. Anticoagulant therapy may substantially decrease the rate of embolic events by 33% compared with no anticoagulation.
Factors contributing to LVMT formation include LV regional-wall akinesia or dyskinesia with blood stasis, injury to and inflammation of the endocardial tissue that provides a thrombogenic surface, and a hypercoagulable state. The most common clinical presentation of patients with LVMT complicating an MI is stroke. Most episodes occur within the first 10 days after AMI. Physical findings depend on the site of embolism.
Transthoracic echocardiography remains the imaging modality of choice and is 92% sensitive and 88% specific for detecting LVMT (see the image below). Management of LVMT includes heparin treatment followed by oral warfarin therapy for 3-6 months. In patients with LVAs, lifelong anticoagulation may be appropriate if a mural clot persists.
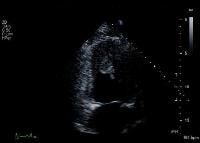
Apical 2-chamber view depicts a large left ventricular apical thrombus with mobile extensions. Pericarditis
The incidence of early pericarditis after MI is approximately 10%, and this complication usually develops within 24-96. Pericarditis is caused by inflammation of pericardial tissue overlying infarcted myocardium. The clinical presentation may include severe chest pain, usually pleuritic, and pericardial friction rub.
The key ECG change is diffuse ST-segment elevation in all or nearly all of leads. Echocardiography may reveal a small pericardial effusion. The mainstay of therapy usually includes aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs). Colchicine may be beneficial in patients with recurrent pericarditis.
Post-MI syndrome (Dressler syndrome)
Before the era of reperfusion, the incidence of post-MI syndrome ranged from 1-5% after AMI, but this rate has dramatically declined with the advent of thrombolysis and coronary angioplasty.
Although the exact mechanism has yet to be elucidated, post-MI syndrome is considered to be an autoimmune process. Clinical features include fever, chest pain, and other signs and symptoms of pericarditis occurring 2-3 weeks after AMI. Management involves hospitalization and observation for any evidence of cardiac tamponade. Treatment comprises rest, use of NSAIDs, and/or steroids in patients with recurrent post-MI syndrome with disabling symptoms.
Previous, Complications of Myocardial Infarction
 This radiograph reveals an enlarged right heart and pulmonary artery dilatation in a 24-year-old woman with an unrestricted patent ductus arteriosus (PDA) and Eisenmenger syndrome.
This radiograph reveals an enlarged right heart and pulmonary artery dilatation in a 24-year-old woman with an unrestricted patent ductus arteriosus (PDA) and Eisenmenger syndrome. 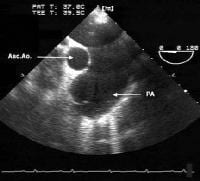 This transesophageal image is from the midesophagus of a patient with Eisenmenger syndrome secondary to an unrestricted patent ductus arteriosus (PDA). It shows a severely dilated pulmonary artery. PA = pulmonary artery, Asc Ao = ascending aorta.
This transesophageal image is from the midesophagus of a patient with Eisenmenger syndrome secondary to an unrestricted patent ductus arteriosus (PDA). It shows a severely dilated pulmonary artery. PA = pulmonary artery, Asc Ao = ascending aorta. 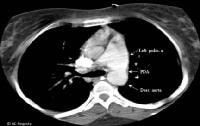 This computed tomography (CT) chest scan shows a large, unrestricted patent ductus arteriosus (PDA) in a 24-year-old woman with Eisenmenger syndrome.
This computed tomography (CT) chest scan shows a large, unrestricted patent ductus arteriosus (PDA) in a 24-year-old woman with Eisenmenger syndrome. 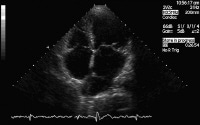 Cor triatriatum. Echocardiogram showing the proximal chamber (PC) and distal chamber (DC) of the left atrium; the right atrium (RA), left ventricle (LV), and right ventricle (RV) also are shown. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy.
Cor triatriatum. Echocardiogram showing the proximal chamber (PC) and distal chamber (DC) of the left atrium; the right atrium (RA), left ventricle (LV), and right ventricle (RV) also are shown. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy. 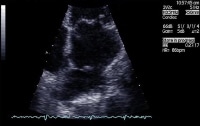 Cor triatriatum. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy.
Cor triatriatum. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy.  Cor triatriatum. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy.
Cor triatriatum. Image courtesy of Guido Giordano, MD, Cardiovascular Department, Azienda Ospedaliera Cannizzaro, Catania, Italy. 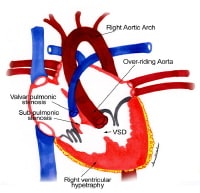 Anatomic findings in tetralogy of Fallot. NextBackground
Anatomic findings in tetralogy of Fallot. NextBackground This image shows completed blocking with a Taussig shunt
This image shows completed blocking with a Taussig shunt 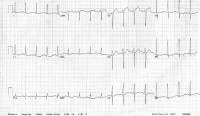 A 62-year-old woman with a history of chronic stable angina and a "valve problem" presents with new chest pain. She is symptomatic on arrival, complaining of shortness of breath and precordial chest tightness. Her initial vital signs are blood pressure = 140/90 mm Hg and heart rate = 98. Her electrocardiogram (ECG) is as shown. She is given nitroglycerin sublingually, and her pressure decreases to 80/palpation. Right ventricular ischemia should be considered in this patient. NextBackground
A 62-year-old woman with a history of chronic stable angina and a "valve problem" presents with new chest pain. She is symptomatic on arrival, complaining of shortness of breath and precordial chest tightness. Her initial vital signs are blood pressure = 140/90 mm Hg and heart rate = 98. Her electrocardiogram (ECG) is as shown. She is given nitroglycerin sublingually, and her pressure decreases to 80/palpation. Right ventricular ischemia should be considered in this patient. NextBackground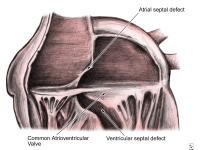 Anatomy of the endocardial cushion defect (ie, complete form); note the common atrioventricular valve straddling the atrial septal and ventricular septal defects.
Anatomy of the endocardial cushion defect (ie, complete form); note the common atrioventricular valve straddling the atrial septal and ventricular septal defects. 













