Torsade de pointes is an uncommon and distinctive form of polymorphic ventricular tachycardia (VT) characterized by a gradual change in the amplitude and twisting of the QRS complexes around the isoelectric line (see the image below). Torsade de pointes, often referred to as torsade, is associated with a prolonged QT interval, which may be congenital or acquired. Torsade usually terminates spontaneously but frequently recurs and may degenerate into ventricular fibrillation.
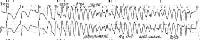 Torsade de pointes. Asymptomatic patient on erythromycin had marked QT prolongation on ECG findings. Patient was profoundly hypomagnesemic and hypokalemic. This shows an example of recurrent nonsustained torsade de pointes that occurred several hours after the ECG was performed. With discontinuation of the erythromycin and aggressive repletion of the magnesium and potassium, no further torsade de pointes occurred and the patient's QT interval returned to normal.
Torsade de pointes. Asymptomatic patient on erythromycin had marked QT prolongation on ECG findings. Patient was profoundly hypomagnesemic and hypokalemic. This shows an example of recurrent nonsustained torsade de pointes that occurred several hours after the ECG was performed. With discontinuation of the erythromycin and aggressive repletion of the magnesium and potassium, no further torsade de pointes occurred and the patient's QT interval returned to normal. In torsade, the morphology of the QRS complexes varies from beat to beat. The ventricular rate can range from 150 beats per minute (bpm) to 250 bpm.
The original report described regular variation of the morphology of the QRS vector from positive to net negative and back again. This was symbolically termed torsade de pointes, or "twisting of the point" about the isoelectric axis, because it reminded the authors of the torsade de pointes movement in ballet. Most cases exhibit polymorphism, but the axis changes may not have regularity.
The definition also requires that the QT interval be increased markedly (usually to 600 msec or greater). Cases of polymorphous ventricular tachycardia in which the QT interval is not prolonged are treated as generic ventricular tachycardia. Torsade usually occurs in bursts that are not sustained; thus, the rhythm strip usually shows the patient's baseline QT prolongation.
The etiology and management of torsade are, in general, quite different from those of garden-variety VT. In particular, the use of group IA antidysrhythmic drugs, which tend to prolong the QT interval, can have disastrous consequences in torsade. Differentiating between these entities, therefore, is critically important.
For more information, go to Ventricular Tachycardia.
NextPathophysiologyThe association between torsade and a prolonged QT interval has long been known, but the mechanisms involved at the cellular and ionic levels have been made clearer in approximately the last decade. The abnormality underlying both acquired and congenital long QT syndromes is in the ionic current flow during repolarization, which affects the QT interval.
A variety of changes in ionic current can result in the common effect of decreased repolarizing current, reflected in a long QT, and these changes can secondarily lead to subsequent depolarizing currents and sometimes action potentials, termed afterdepolarizations. This leads to a further delay in repolarization and causes early afterdepolarization (EAD), the triggering event for torsade.
Repolarization has 3 phases. During the initial upstroke of action potential in a normal cardiac cell, a rapid net influx of positive ions (Na+ and Ca++) occurs, which results in the depolarization of the cell membrane. This is followed by a rapid, transient outward potassium current (Ito), while the influx rate of positive ions (Na+, Ca++) declines. This represents the initial part of the repolarization, or phase 1.
Phase 2 is characterized by the plateau. The positive currents flowing inward and outward become almost equal during this stage.
Phase 3 of repolarization is mediated by activation of the delayed rectifier potassium current (IK) moving outward while the inward positive current decays. If a slow inactivation of the Ca++ and Na+ currents occurs, this inward "window" current can cause single or repetitive depolarization during phases 2 and 3 (ie, EADs). These EADs appear as pathologic U waves on a surface ECG, and, when they reach a threshold, they may trigger ventricular tachyarrhythmias.
These changes in repolarization do not occur in all myocardial cells. The deep endocardial region and midmyocardial layer (composed of M cells) of the ventricle are more prone to prolongation of repolarization and EADs because they have a less-rapid delayed rectifier potassium current (IKr), while other regions might have short or normal cycles. This heterogeneity of repolarization in the myocardial cells promotes the spread of triggered activity, which is initiated by EADs by a reentrant mechanism and currently is thought to be responsible for the maintenance of torsade.[1]
Six genetic variants underlying torsade are currently recognized. Genotypes LQT1 and LQT2 have slow potassium channels, while LQT3 shows defects in the sodium channels. Treatment modalities soon may be based on the genotype of the individual.
PreviousNextEpidemiology of TorsadeThe prevalence of torsade is unknown. Torsade is a life-threatening arrhythmia and may present as sudden cardiac death in patients with structurally normal hearts.[2] In the United States, 300,000 sudden cardiac deaths occur per year. Torsade probably accounts for fewer than 5%.
For both sexes, the corrected QT interval is longer in white persons than in black persons, possibly explaining the lower susceptibility to acquired torsade in black persons. Brugada syndrome is more frequent in Southeast Asians.[3]
Torsade is 2-3 times more common in women than in men. Women have longer QT intervals,[4] and have more QT prolongation secondary to drug therapy. Congenital long QT syndrome is autosomal dominant but shows greater frequency of expression and a greater lengthening of the QT interval in women than in men.
Torsade occurs in patients of a wide age range, from newborns to the very elderly. The highest frequency is in patients aged 35-50 years. Torsade that occurs at an early age usually is due to congenital long QT syndrome. In older persons, it usually is due to acquired long QT syndrome.
PreviousNextClinical PresentationPatient historyPatients with torsade usually present with recurrent episodes of palpitations, dizziness, and syncope; however, sudden cardiac death can occur with the first episode. Nausea, cold sweats, shortness of breath, and chest pain also may occur but are nonspecific and can be produced by any form of tachyarrhythmia.
In a young patient with torsade, a diagnosis of congenital long QT syndrome should be considered, especially if a family history of sudden cardiac death or sudden infant death syndrome is present. In these patients, episodes of torsade are triggered by adrenergic stimulation such as stress, fear, or physical exertion, but other predisposing factors also should be considered. See Long QT Syndrome.
A family history of congenital deafness may also be suggestive, although a prolonged QT is found in only 0.25-0.3% of deaf-mute children. Patients with Jervell and Lange-Nielsen syndrome commonly have congenital sensorineural deafness representing an autosomal dominant pattern of inheritance for cardiac abnormalities, whereas deafness usually is autosomal recessive.
Another form of familial or congenital long QT syndrome is Romano-Ward syndrome, in which hearing is normal and an autosomal dominant pattern of inheritance is observed.
Patients with acquired long QT syndrome usually develop torsade during periods of bradycardia. The most common causes of acquired long QT syndrome are medications and electrolyte disorders (eg, hypokalemia, hypomagnesemia). Drug-associated torsade de pointes is relatively rare, but is becoming increasingly common; its incidence is as high as 2-3% with certain drugs. Hence, asking the patient about all current medications is important.
Physical examinationThe physical findings in torsade depend on the rate and duration of tachycardia and the degree of cerebral hypoperfusion.
Findings include rapid pulse, low or normal blood pressure, or transient or prolonged loss of consciousness. This could be preceded by bradycardia or premature ventricular contractions (leading palpitations).
Pallor and diaphoresis may be noted, especially with a sustained episode.
Other physical signs depend on the etiology of torsade.
Risk factorsRisk factors for torsade include the following:
Congenital long QT syndromeFemale genderAcquired long QT syndrome (causes of which include medications and electrolyte disorders such as hypokalemia and hypomagnesemia)BradycardiaBaseline electrocardiographic abnormalitiesRenal or liver failurePreviousNextEtiology of TorsadeProlongation of the QT interval may be congenital, as seen in the Jervell and Lange-Nielsen syndrome (ie, congenitally long QT associated with congenital deafness) and the Romano Ward syndrome (ie, isolated prolongation of QT interval). Both of these syndromes are associated with sudden death due to either primary ventricular fibrillation or torsade that degenerates into ventricular fibrillation.
Brugada syndrome is characterized by a coved ST segment in the right precordial leads. The syndrome may cause sudden death due to polymorphic VT resembling torsade.
Takotsubo cardiomyopathy (stress-induced cardiomyopathy) causes a predisposition to torsade.[5]
The acquired conditions that predispose one to torsade either decrease the outward potassium current or interfere with the inward sodium and calcium currents, or fluxes.
The electrolyte disturbances that have been reported to precipitate torsade include hypokalemia and hypomagnesemia. These disturbances cause a delay in phase III (ie, reprolongation) and form the substrate for emergence of the dysrhythmia. Close observation is required in predisposed patients, such as those with cirrhosis or hypothyroidism.
Antiarrhythmic drugs reported to be etiologic include class IA agents (eg, quinidine, procainamide, disopyramide), class IC agents (eg, encainide, flecainide), and class III agents (eg, sotalol, amiodarone).
Drug interactions with the antihistamines astemizole (recalled from US market) and terfenadine (recalled from US market) can precipitate torsade; these drugs should never be used with class IA, IC, or III agents. Astemizole and terfenadine, in high dosages or when used in combination with the azole antifungal drugs or the macrolide antibiotics, have been reported to precipitate torsade and sudden death.
Grapefruit juice has been shown to slow the hepatic metabolism of these antihistamines as well as other drugs and to prolong the QT interval in patients taking astemizole or terfenadine. Clinical implications of this interaction are unclear.
Other drugs that prolong the QT interval and have been implicated in cases of torsade include phenothiazines, tricyclic antidepressants, lithium carbonate, ziprasidone,[6] cisapride, highly active antiretroviral drugs, high-dose methadone, anthracycline chemotherapeutic agents (eg, doxorubicin, daunomycin), some fluoroquinolones, and any other medication using the CYP3A metabolic pathway. Ranolazine,[7] an antiangina agent, also prolongs the QTc, but torsade is a rare complication of this therapy.
Congenital long QT syndromes (adrenergic-dependent)The following congenital syndromes are associated with torsade:
Jervell and Lange-Nielsen syndromeRomano-Ward syndromeAcquired long QT syndromesDrugs in a number of drug classes have been associated with torsade.
Antiarrhythmic drugs associated with torsade include the following:
Class IA - Quinidine, disopyramide, procainamideClass III - Sotalol, amiodarone (rare), ibutilide, dofetilide, almokalantOther drug classes associated with torsade include the following:
Antibiotics - Erythromycin, clarithromycin, azithromycin, levofloxacin, moxifloxacin, gatifloxacin, trimethoprim-sulfamethoxazole, clindamycin, pentamidine, chloroquine Antifungals - Ketoconazole, itraconazoleAntivirals – AmantadineAntipsychotics - Haloperidol, phenothiazines, thioridazine, trifluoperazine, sertindole, zimeldine, ziprasidone[8] Tricyclic and tetracyclic antidepressantsAntihistamines (histamine1-receptor antagonists) - Terfenadine, astemizole, diphenhydramine, hydroxyzineCholinergic antagonists - Cisapride, organophosphates (pesticides)Diuretics - Indapamide, hydrochlorothiazide, furosemideAntihypertensives - Bepridil, lidoflazine, prenylamine, ketanserinLithiumAnticonvulsants - phenytoin, carbamazepine (possible)Oral hypoglycemicCitrate (massive blood transfusions)CocaineVasopressin (possible)Fluoxetine (possible)Some drugs (eg, amiodarone) routinely prolong QT but are less commonly associated with clinical consequences of long QT. For additional information, see QTsyndrome.ch.
Conditions associated with torsade include the following:
Electrolyte abnormalities - Hypokalemia, hypomagnesemia, hypocalcemiaEndocrine disorders - Hypothyroidism, hyperparathyroidism, pheochromocytoma, hyperaldosteronismCardiac conditions - Myocardial ischemia, myocardial infarction, myocarditis, bradyarrhythmia, complete atrioventricular (AV) block, takotsubo cardiomyopathyIntracranial disorders - Subarachnoid hemorrhage, thalamic hematoma, cerebrovascular accident, encephalitis, head injuryNutritional disorders - Anorexia nervosa, starvation, liquid protein diets, gastroplasty and ileojejunal bypass, celiac diseasePreviousNextDifferential Diagnosis for TorsadeThe differential diagnosis includes the following:
Ventricular TachycardiaPediatrics, TachycardiaSyncopeRenal Failure, Chronic and Dialysis ComplicationToxicity, AntidysrhythmicToxicity, AntihistamineVentricular FibrillationSudden Cardiac DeathOther considerations are the differentiation of acquired long QT syndrome from congenital long QT syndrome. In addition, torsade should be differentiated from polymorphic ventricular tachycardia or, rarely, monomorphic ventricular tachycardia.
Supraventricular tachycardia with aberrant conduction may be confused with torsade. One clue to the former is that atrial fibrillation may be intermixed with narrower and typical QRS complexes.
PreviousNextElectrocardiographyTorsade is an electrocardiographic diagnosis, and obtaining an ECG is mandatory.[9] Typical examples are depicted below.
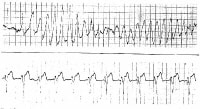 A run of torsade de pointes in a 70-year-old man who developed QT prolongation (QTc = 0.61 sec) secondary to quinidine therapy. The bottom strip shows resolution with overdrive ventricular pacing.
A run of torsade de pointes in a 70-year-old man who developed QT prolongation (QTc = 0.61 sec) secondary to quinidine therapy. The bottom strip shows resolution with overdrive ventricular pacing. 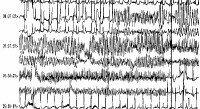 A patient with prolonged QT and atrial ectopy. Strip shows premature beat that entrains and a run of torsade de pointes.
A patient with prolonged QT and atrial ectopy. Strip shows premature beat that entrains and a run of torsade de pointes. Frequent ECG monitoring is indicated for patients who are at risk due to chronic conditions or drug therapy. When the patient is in sinus rhythm, examine the QT interval. Usually, a prolonged QT interval and pathological U waves are present, reflecting abnormal ventricular repolarization. The most consistent indicator of QT prolongation is a QT of 0.60 s or longer or a QTc (corrected for heart rate) of 0.45 s or longer.
Other electrocardiographic features helpful in diagnosing torsade include its typical mode of onset and its morphology, as follows:
Patients have paroxysms of 5-20 beats at a rate faster than 200 bpm; sustained episodes occasionally can be seenProgressive change in polarity of QRS about the isoelectric line occursComplete 180° twist of QRS complexes in 10-12 beats is presentA short-long-short sequence between the R-R interval occurs before the trigger response.Patients may revert spontaneously or convert to a nonpolymorphic ventricular tachycardia or ventricular fibrillationOccasionally, T-wave alternans may be seen before torsadeTorsade occurring in the setting of acquired long QT syndrome is preceded by pauses in almost all cases. In congenital long QT syndrome (adrenergic-dependent), pause dependence is found in most of the adult cases, whereas onset of torsade is not pause-dependent in children.
Failure to identify this rhythm may occur for various reasons. During very short runs of torsade, the typical twisting of the QRS complexes around the isoelectric line may not be apparent. Early events usually are short-lived. In the case of a single-lead recording, the typical morphology of torsade may not be obvious.
The diagnosis of torsade should be considered in any patient with pause-dependent ventricular tachycardia, and ventricular bigeminy in a patient with long QT interval may be a sign of an impending torsade.
Findings from electrophysiological studies usually are negative in torsade.
PreviousNextLaboratory StudiesElectrolytesCheck for hypokalemia, hypomagnesemia, and hypocalcemia.
Cardiac enzymesRule out myocardial ischemia, especially in patients without QT prolongation.
Imaging studiesChest radiographs and echocardiography should be performed to rule out structural heart disease, if any clinical suggestion is present.
Other testsOther tests should be ordered depending on the etiological factors being considered (see Etiology of Torsade).
PreviousNextTreatment of TorsadeAcute managementTreatment can be divided into short-term and long-term management. Short-term management of torsade is the same in both acquired and congenital long QT syndrome, except that beta1-adrenergic stimulation may be tried in the acquired form but is contraindicated in the congenital form.
In an otherwise stable patient, DC cardioversion is kept as a last resort because torsade is paroxysmal in nature and is characterized by its frequent recurrences following cardioversion. Although torsade frequently is self-terminating, it may degenerate into ventricular fibrillation, which requires direct current (DC) defibrillation.
Any offending agent should be withdrawn. Predisposing conditions such as hypokalemia, hypomagnesemia, and bradycardia should be identified and corrected.
Pharmacologic therapyMagnesium is the drug of choice for suppressing EADs and terminating the arrhythmia. Magnesium achieves this by decreasing the influx of calcium, thus lowering the amplitude of EADs.
Magnesium can be given at 1-2 g IV initially in 30-60 seconds, which then can be repeated in 5-15 minutes. Alternatively, a continuous infusion can be started at a rate of 3-10 mg/min. Magnesium is effective even in patients with normal magnesium levels. Because of the danger of hypermagnesemia (depression of neuromuscular function), the patient requires close monitoring.
Some authorities recommend supplemental potassium to increase the potassium concentration to high normal, which increases the efflux of potassium from myocardial cells, thus causing rapid repolarization.
Lidocaine usually has no effect in torsade. Occasionally, it can have an initial beneficial effect, but torsade recurs in all cases.
Mexiletine also may be helpful in suppressing torsade. In one study, it was used in patients with HIV who had acquired long QT interval and torsade. It effectively suppressed the torsade on a long-term basis.
Patients with congenital long QT syndromes are thought to have an abnormality of sympathetic balance or tone and are treated with beta-blockers. If the patient experiences breakthrough torsade, a short-acting beta-blocker, such as esmolol, can be tried.[10]
Isoproterenol can be used in bradycardia-dependent torsade that usually is associated with acquired long QT syndrome (pause-dependent). It should be administered as a continuous IV infusion to keep the heart rate above 90 bpm.
Isoproterenol accelerates AV conduction and decreases the QT interval by increasing the heart rate and reducing temporal dispersion of repolarization. Beta-adrenergic agonists such as isoproterenol are contraindicated in the congenital form of long QT syndrome (adrenergic-dependent). Because of precautions, contraindications, and adverse effects associated with its use, this drug is used as an interim agent until overdrive pacing can be started.
Temporary transvenous pacingBased on the fact that the QT interval shortens with a faster heart rate, pacing can be effective in terminating torsade. It is effective in both forms of the long QT syndrome because it facilitates the repolarizing potassium currents and prevents long pauses, suppressing EADs and decreasing the QT interval.
Atrial pacing is the preferred mode because it preserves the atrial contribution to ventricular filling and also results in a narrower QRS complex and hence a shorter QT. In patients with AV block, ventricular pacing can be used to suppress torsade.
Pacing should be instituted at a rate of 90-110 bpm until the QT interval is normalized. Overdrive pacing may be necessary at a rate of up to 140 bpm to control the rhythm.
The patient with torsade who is in extremis should be treated with electrical cardioversion or defibrillation. Anecdotal reports cite successful conversion with phenytoin (Dilantin) and lidocaine. A few cases of successful conversion using phenytoin and overdrive pacing have been reported.
If patient is unresponsive to conversion with phenytoin and overdrive pacing, attempt electrical cardioversion.
Long-term treatmentBeta-adrenergic antagonists at maximally tolerated doses are used as a first-line long-term therapy in congenital long QT syndrome. Propranolol is used most extensively, but other agents such as esmolol or nadolol also can be used. Beta-blockers should be avoided in those congenital cases in which bradycardia is a prominent feature. Beta-blockers are contraindicated in acquired long QT syndrome because bradycardia produced by these agents can precipitate torsade.
Patients without syncope, ventricular tachyarrhythmia, or a family history of sudden cardiac death can be observed without starting any treatment.
Permanent pacing benefits patients who remain symptomatic despite receiving the maximally tolerated dose of beta-blockers and can be used adjunctively with beta-blockers. It decreases the QT interval by enhancing the repolarizing potassium currents and suppressing EADs.
High left thoracic sympathectomy, another antiadrenergic therapy, is effective in patients who remain refractory to beta-blockade and pacing. Accidental ablation of ocular efferent sympathetic nerves may result in Horner syndrome.
Implantable cardioverter-defibrillators (ICDs) are useful in instances when torsade recurs despite treatment with beta-blockers, pacing, and possibly left thoracic sympathectomy. Beta-blockers should be used along with ICDs because shock can further precipitate torsade by adrenergic stimulation. In the United States, an ICD for refractory cases may often precede sympathectomy.
Long-term treatment in acquired long QT syndrome usually is not required because the QT interval returns to normal once the inciting factor or predisposing condition has been corrected. Pacemaker implantation is effective in cases that are associated with heart block or bradycardia. ICDs are indicated in cases that cannot be managed by avoidance of the offending agent.
The boundary between acquired and congenital may not always be clear. Additive factors are often present, and individuals may show increased susceptibility to QT effects.
ConsultationsImmediate cardiology evaluation and follow-up are required. Other possible consultations include the following:
ElectrophysiologistCardiologistGeneticist (in cases of familial or congenital long QT syndrome)ActivityCompetitive sports are prohibited in patients with congenital long QT syndrome.
ComplicationsComplications may include the following:
Monomorphic ventricular tachycardiaVentricular fibrillationSudden cardiac deathPrognosisIn congenital long QT syndrome, the mortality rate for untreated patients is 50% in 10 years, which can be reduced to 3-4% with therapeutic intervention.
In acquired long QT syndrome, the prognosis is excellent once the inciting factor has been identified and reliably withheld.
Patient educationInstruct patients to use medications only with the approval of a physician. Instruct patients to avoid competitive sports (in cases of congenital long QT syndrome).
Close follow-up is needed because of a risk of sudden cardiac death. Offer emotional support; suggest attending a cardiac support group.
Patients should be taught how to monitor their pulse and recognize adverse drug effects. Families should undergo training for basic life support.
Previous, Torsade de Pointes












0 comments:
Post a Comment